Applications
Please, contact us to discuss how we can be of assistance in achieving your project goals or to receive a quote for your project.

de novo Genome Assembly
De novo genome assembly can be done using several strategies, depending on the genome size, number of samples and desired completeness of the genome.
-
PacBio REVIO: This is the latest instrument from PacBio, it produces long reads with high accuracy. It is the technology of choice for assembly of eukaryotic and bacterial genomes. Also, it is used for characterization of full-length 16S and other amplicons and of full-length transcriptomes (IsoSeq). One REVIO Cell produces up to 120 Gbases of HiFi error-corrected reads with fragments 10-30kb, or up to 100 million error-corrected cDNA or amplicon reads.
-
Illumina Novaseq X Plus: used for scaffolding existing assemblies by sequencing Hi-C and/or TellSeq libraries, see 4 and 5.
-
Oxford Nanopore GridION: this technology typically produces reads 5kb to 30kb and longer if desired. It can be used for assembly of microbial and fungal genomes and for scaffolding of eukaryotic genomes.
-
Tell-Seq libraries: this technology enables the fast and low scaffolding of genomes by barcoding long molecules of DNA. They enable estimation of genome size and complexity with Genome Scope and scaffolding of Pacbio assemblies.
-
Hi-C libraries: these are long-range Hi-C libraries used for scaffolding of existing assemblies.

Genome Resequencing and skim-seq
This application involves the creation of PCR-free or shotgun libraries using 3-4 cycles of PCR when sufficient DNA is unavailable. These libraries are barcoded with Unique Dual Indexes (UDIs) to prevent index switching and contain fragments with an average length of 450 bp. Each sample is sequenced to the desired depth (ranging from 1x to 100x or more) on either a NovaSeq or MiSeq, depending on genome size, the number of samples, and desired genome coverage. Skim-seq libraries are less efficient at amplifying the entire genome but offer significant cost savings, allowing for genotyping of a large number of samples at a reasonable expense.
An exciting new application involves resequencing genomes using Long Reads (Pacbio Revio), which is significantly more cost-effective. This approach not only detects SNPs and small indels but also identifies larger structural variants and translocations that are often absent from short-read data.
RNA-Seq and Small RNA
There are several options for RNA-Seq and small RNA libraries:
RNA-Seq, Eukaryotic species:
mRNA-enriched: these libraries are constructed by first selecting polyA+ mRNAs, converting the mRNAs to cDNA, performing adaptor ligation and amplifying for the minimum number of PCR cycles required. At least 50ng of total RNA having a RIN > 7 and free from contaminating DNA should be submitted.
rRNA-depleted: for characterization of polyA+ and polyA- transcripts, libraries can be constructed by depleting rRNA and converting the leftover RNA to cDNA, instead of capturing polyA+ mRNAs.
FFPE samples: RNA-Seq libraries can be constructed from degraded and very low amounts of RNA by first converting all the RNA to cDNA and then using specific probes to remove rRNA, or by using probes that capture specific transcripts.
IsoSeq = full-length polyA mRNAs are sequenced in the PacBio Revio with high accuracy. This is the best way to fully characterize an entire transcriptome, including all splice-variants.
RNAseq, Microbial and Metagenomic samples:
rRNA-depletion: removal of rRNA can be done with probes that recognize microbial rRNAs or with probes that remove both host and microbial rRNAs. The leftover RNA is converted to RNA-Seq libraries that are individually barcoded with Unique Dual Indexes.
Small RNA, Eukaryotic species:
Libraries are constructed from 10ng of enriched small RNA fraction or 100ng of total RNA. RNAs from 15nt to 30nt in length are enriched by size selection on a PAGE gel.
Small RNA, bacterial:
Total RNA is treated with Antarctic Phosphatase and PNK, adaptors are added and RNAs 15nt to 250nt are enriched by size selection on a PAGE gel.
Circular RNAs, double-stranded RNAs:
We have extensive experience in the construction of these libraries.
Epigenetics
Methylation Sequencing:
Whole genome methylome analysis involves an enzymatic treatment that identifies two types of DNA modifications: 5-methylcytosine (5-mC) and 5-hydroxymethylcytosine (5-hmC). Alternatively, reduced-representation bisulfite sequencing libraries (RRBS) can be constructed from areas of the genome that have high CpG content. Whole genome methylome libraries are typically sequenced to a depth of 10X to 30X, while RRBS libraries typically capture only 1% of the genome so they need a much lower sequencing depth.
ATAC-Seq, Cut&Run:
We have extensive experience in the construction of these libraries. If you want to make them in your lab, we recommend discussing your application with us prior to library construction to ensure that final libraries will be compatible with the latest sequencing technology.
The critical parameters for these libraries are:
- The input DNA must be sheared to a size between 100bp and 600bp prior to pull down. We get consistent results using Covaris sonicators. Ideal sonication times need to be validated for different cell types and range from 10 min to 35 min. After sonication of a test cell suspension, please purify an aliquot of the cells and run the DNA on a 1% or 2% agarose gel with appropriately sized DNA ladder to confirm that the DNA is between 100bp and 600bp. Longer DNAs do not sequence well are most likely not efficiently enriched.
- After immunoprecipitation, we strongly recommend to confirm enrichment by performing qPCR of positive control regions relative to negative control regions before submitting DNA for library construction. A collection of ChIP-Seq data is available at the encode webpage, where the peaks can be visualized on the genome browser and primers can be designed from your genomic regions of interest. A database of primers for many common targets can be found here.


Long Reads for de novo Assembly and Genome Phasing
There are three technologies available in our facility for sequencing and assembly of long reads and for genome phasing:
- PacBio REVIO: This is the latest instrument from PacBio, it produces long reads with high accuracy. It is the instrument of choice for assembly of complex genomes. Also, it is used for characterization of full-length 16S and other amplicons and of full-length transcriptomes (IsoSeq). One REVIO Cell produces 120 Gbases of HiFi error-corrected reads with fragments 10-30kb, or ~ 10-13 million error corrected cDNA or amplicon reads.
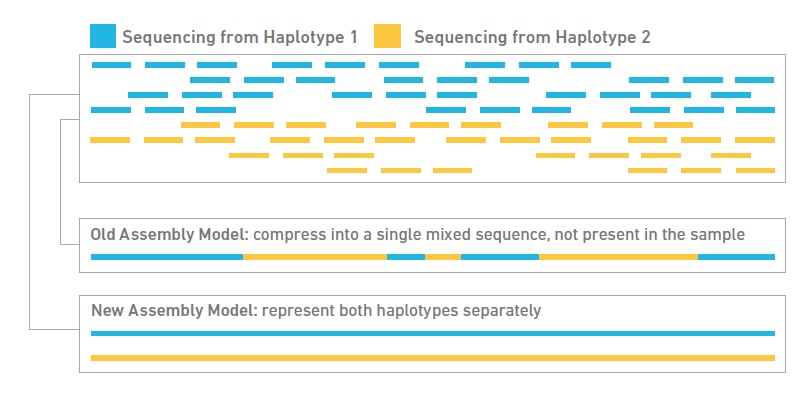
- Tell-Seq libraries for de novo assembly and/or phasing: This technology begins with high molecular weight (HMW) DNA, ideally in fragments at least 50kb or greater. It then partitions the HMW DNA fragments into micelles, along with an adapter molecule and a barcode. All the DNA fragments within an individual micelle get barcoded with the same barcode. Fragments are then pooled and converted into a standard shotgun library and sequenced on an Illumina NovaSeq. The data can be used for genome scaffolding, phasing and discovery of short and large structural variants, including SNPs.
- Oxford Nanopore GridION x5: This technology typically produces reads 10kb to 50kb and longer if desired.
Single Cell Transcriptomics and Spatial Transcriptomics
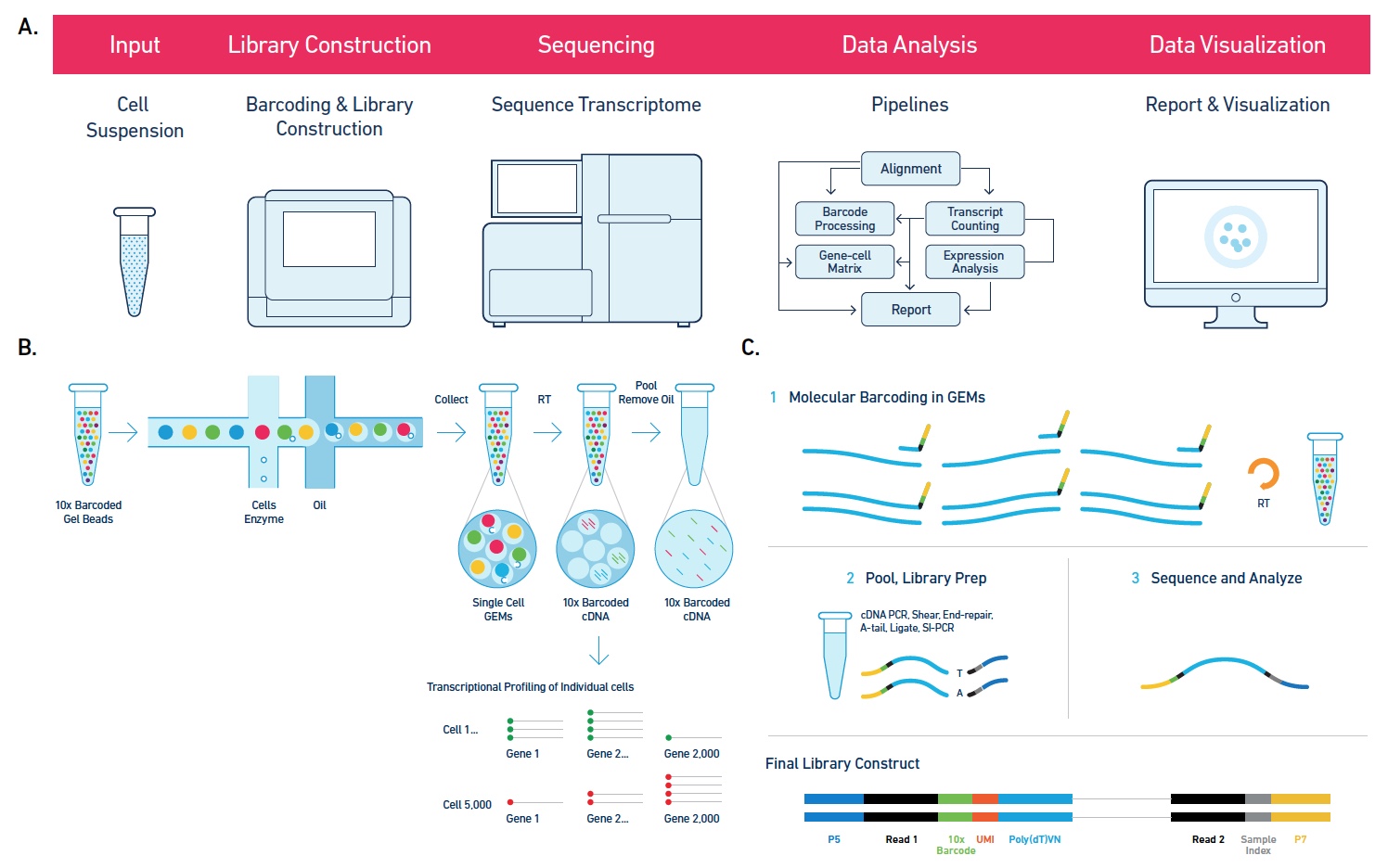
Single Cell 10x Genomics: Single cells or nuclei from any eukaryotic species can be processed with this technology, as long as the cells are < 40uM in diameter. Libraries for RNASeq, ATAC, or Multiome (RNASeq or ATAC) can be constructed, as well as probe-based Fixed RNA libraries for human and mouse samples. This technology begins with either a single cell or nuclei suspension, with > 70% viability for cells. It then partitions the single cells into micelles, along with an adapter molecule and a barcode enclosed within a gel bead. All the mRNAs from a single-cell within an individual micelle get barcoded with the same barcode. mRNAs are then converted to cDNAs, pooled, converted into an Illumina library and sequenced on a NovaSeq to get at least 50,000 reads per cell. Sequencing requires 28 cycles for read1 (barcode) and at least 90 cycles for read2 (cDNA). Typically this is performed on a 2x100nt or 2x150nt paired-end lane. Read more about what you can do with 10x Genomics here.
The software Cell Ranger is utilized to align the reads to the appropriate annotated genome/transcriptome and generate gene counts and other features.
Please see the 10x Submission Guidelines for important submission information, protocols, and tips.
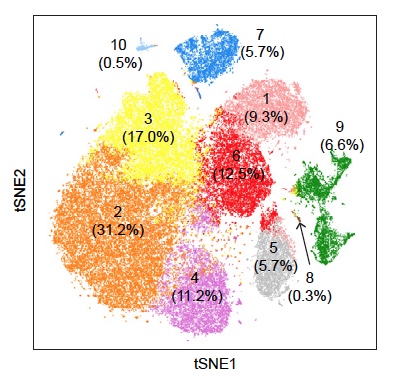
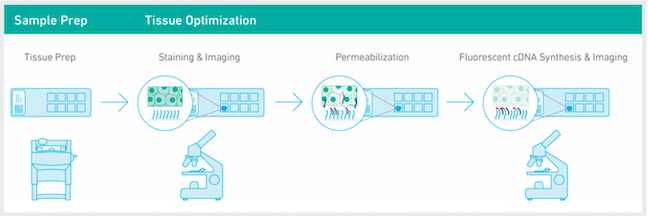
10x Visium Spatial Transcriptomics: The 10x Visium Spatial platform allows researchers to generate spatially resolved gene expression data directly from fresh frozen tissue sections. Tissue sections are first H&E or IF stained and images captured, then the same tissue section is permeabilized to release mRNA onto capture spots that contain spatially barcoded oligos fixed to the slide. mRNAs are converted to cDNAs and then collected for dual-indexed Illumina library construction and sequencing. The H&E stained image and the spatially barcoded cDNAs are overlaid to allow visualization of the gene expression within the original tissue placement. Libraries can also be made from FFPE samples using the FFPE Visium kit.
Please see the 10x Visium page for important submission information, protocols, and tips.
Genotyping
Targeted genotyping, using primers to amplify specific regions of the genome, is accomplished in our facility using the Fluidigm system, a streamlined and repeatable micro-fluidics process that minimizes PCR errors and cross-contamination. This system allows for processing up to 1,500 different samples with up to 48 primer pairs targeting known regions of the genome. We also have extensive experience sequencing GBS and RAD-Seq libraries prepared by our users.
Skim-seq libraries and sequencing is another cost effective way to sequence entire genomes for genotyping at a reasonable cost.
Metagenomics/16S
There are several approaches for the characterization of microbial communities to answer the questions "who is there?", "what are they doing?" and "what are they doing right now?". These involve strategies to characterize the microbes present in the community, the genes that are present in the community, and the genes that are being expressed under different conditions.
-
Whole Genome Sequencing: preparation of shotgun libraries and deep sequencing. The depth of sequencing depends on the expected diversity of the community. We offer low cost methods for the processing of hundreds or thousands of samples and sequencing on the NovaSeq or MiSeq.
-
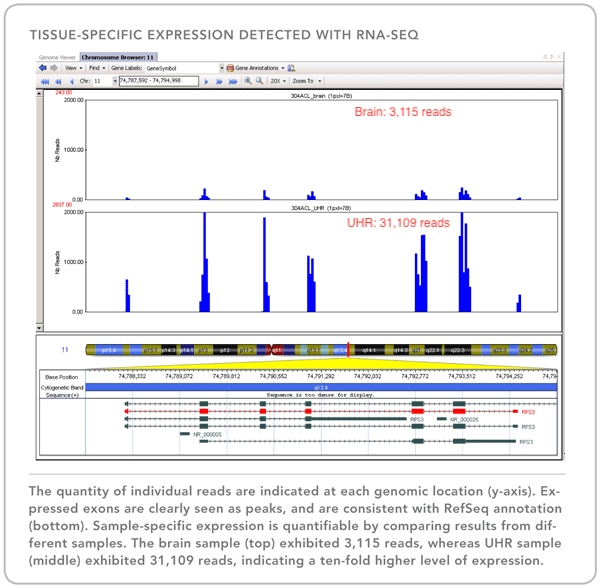
16S rDNA regions and other loci: Our combined Fluidigm+MiSeq protocol is a cost-effective system allowing researchers to sequence PCR products from up to 1500 different metagenomic samples utilizing up to 24 different genes/targets (eg: 16S for bacteria, 16S for archaea, ITS, 18S, and functional genes of interest). We use a streamlined and repeatable micro-fluidics process that minimizes PCR errors and cross-contamination while maximizing your potential understanding of the mixed communities being studied (see Figure 2 below).
-
**Full-length 16S and other amplicons with PacBio**: amplification of the entire 16S gene with the primers AGRGTTYGATYMTGGCTCAG (forward) and RGYTACCTTGTTACGACTT (reverse) produces full length 16S amplicons for sequencing on the PacBio. These amplicons are sequenced with the HiFi mode, which produces amplicons with 99.999% accuracy. Analysis of full-length amplicons produces a more accurate view of the metagenomics community and higher % of classifications to the species level compared to sequencing 16S variable regions.
-
Metatranscriptomics: expressed genes from a metagenomics community can be characterized with RNA-Seq libraries. For this, rRNAs from microbes or from microbes+host are removed with Ribozero Plus or custom probes and RNA-Seq libraries are constructed from the leftover RNA, which contains the expressed genes.
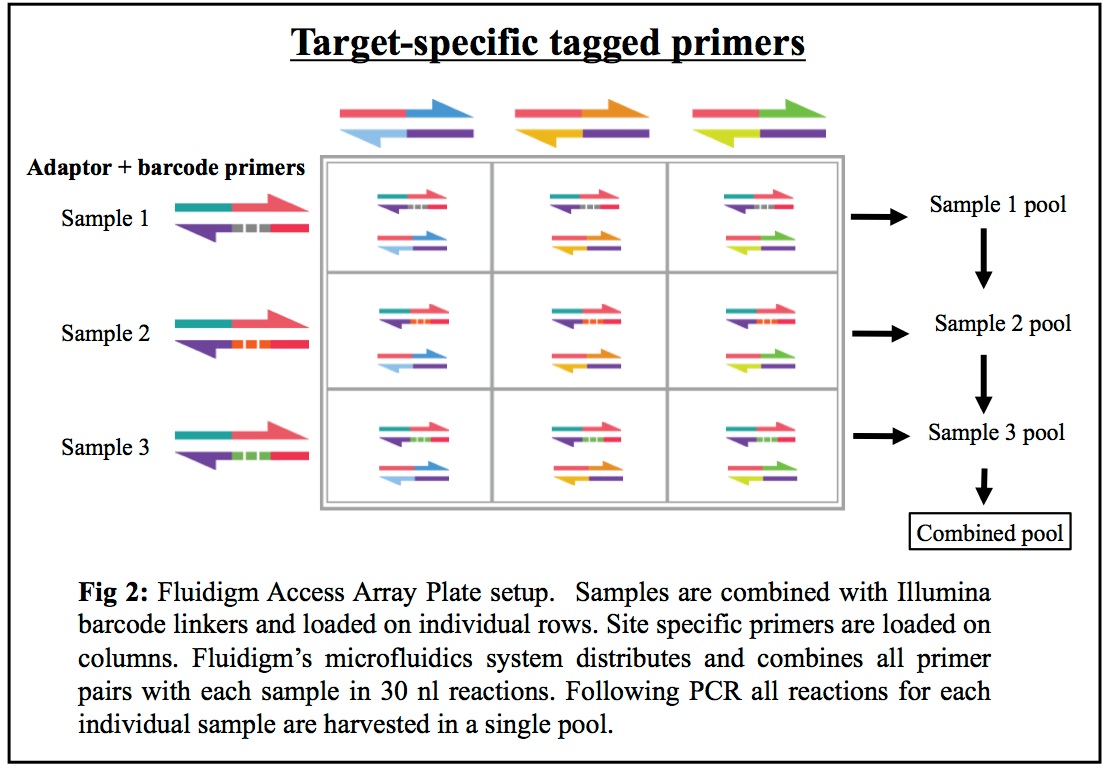
Common Primers Offered for Amplicon Sequencing of 16S regions and other targets
We offer hundreds of different PCR primers for 16S rRNA as well as 18S, eukaryotic, ITS, archaeal, and other functional gene targets. Our list is continuously being expanded, so please check with us for the latest primer selections.
|
Target |
Primer Name |
Primer Sequence (5' to 3') |
Expected FINAL* |
|
16S V1-V3 |
V1-V3 F28 |
GAGTTTGATCNTGGCTCAG |
643 |
|
|
V1-V3 R519 |
GTNTTACNGCGGCKGCTG |
|
|
16S V3-V5 |
V3-V5 F357 |
CCTACGGGAGGCAGCAG |
694 |
|
|
V3-V5 R926 |
CCGTCAATTCMTTTRAGT |
|
|
16S V4 |
V4 515F |
GTGCCAGCMGCCGCGGTAA |
252 |
|
|
V4 806R |
GGACTACHVGGGTWTCTAAT |
|
| 16S V4 (new) | V4 515F (new) | GTGYCAGCMGCCGCGGTAA | 252 |
| V4 806R (new) | GGACTACNVGGGTWTCTAAT | ||
|
Archaea |
Arch349F |
GYGCASCAGKCGMGAAW |
528 |
|
|
Arch806R |
GGACTACVSGGGTATCTAAT |
|
|
Eukaryotic 18S |
F566Euk |
CAGCAGCCGCGGTAATTCC |
765+ |
|
|
R1200Euk |
CCCGTGTTGAGTCAAATTAAGC |
|
|
Eukaryotic 18S |
Euk_1391F |
GTACACACCGCCCGTC |
200-280 |
|
|
EukBr-7R |
TGATCCTTCTGCAGGTTCACCTAC |
|
|
ITS1-ITS4 |
ITS1 |
TCCGTAGGTGAACCTGCGG |
580+ |
|
|
ITS4R |
TCCTCCGCTTATTGATATGC |
|
|
ITS3-ITS4 |
ITS3F |
GCATCGATGAAGAACGCAGC |
462+ |
|
|
ITS4R |
TCCTCCGCTTATTGATATGC |
|
How to Submit Samples for amplification and sequencing on the Fluidigm+MiSeq/NovaSeq:
The success of the amplicon construction and sequencing depends heavily on the integrity and purity of the DNA. Degraded or fragmented DNA produces weak amplicons with amplification artifacts. Even if just one of the DNA samples is fragmented, the artifacts produced by amplification can affect the results of the entire pool. DNA with humic acids or other contaminants that interfere with amplification will also produce poor results. Removal of RNA during DNA purification is preferred.
Please, check as many of your DNA samples as possible on a 1% gel to evaluate the integrity of the DNA:
Run an aliquot (~50 to 100ng) of the DNA on a 1% agarose gel next to an appropriate DNA ladder. This picture can be used to evaluate integrity of the DNA as well as presence/absence of RNA.
For Fluidigm submission of metagenomic/16S assays we require at least 2 ng/ul quantitated by Qubit or Picogreen. Submit at least 10 ul of each DNA sample. Please contact our sister unit, Functional Genomics, for the latest primer information, submission guidelines and submission form.
The DNA Services lab is available to discuss all sequencing options and pricing (contact information below).

Exome and other Targeted Sequencing
Specific regions of the genome, such as exomes or other regions of interest, can be captured by shearing genomic DNA and pulling down those regions of interest with biotinylated probes. The enriched fragments are then adaptored to construct individually barcoded Illumina libraries. Genomic probes can be synthesized for any species with a draft reference genome.
 Core Fragment Analysis and Sanger Sequencing
Core Fragment Analysis and Sanger Sequencing
Please visit our CoreLims website (unicorn.biotec.illinois.edu) to submit your fragment analysis orders and low- and medium-throughtput Sanger sequencing orders.
The Core Facility provides Sanger DNA sequencing and fragment analysis services to researchers at domestic and international academic institutions, government agencies and private companies. All sequencing and fragment analysis samples are analyzed on two Applied Biosystems 3730xl DNA Analyzers. These automated sequencers run with 50cm capillary arrays and are extremely fast and accurate.
We offer two levels of pricing, based on the number of samples in your project and the way you submit them.
- Low-throughput: 1-95 sequencing reactions submitted at any one time in 1.5 ml tubes.
- High-throughput: 1 or more 96-well plates of sequencing reactions submitted at any one time.
Our CoreLims website (unicorn.biotec.illinois.edu) is used to submit and track Fragment Analysis and Sanger sequencing orders, retrieve data, gain access to analysis software and obtain troubleshooting assistance.
Customers are encouraged to discuss any problems or concerns with facility personnel, as our goal is to provide only the highest-quality sequencing data and support. For consultations, please contact the Core staff at dna-seq@illinois.edu or 217-333-9520.